![]()
About AliTV¶
AliTV (Alignment Toolbox & Visualization) is a free software for visualizing whole genome alignments as circular or linear maps. The figure is presented on a HTML-page with an easy-to-use graphical user interface. Comparisons between multiple genomic regions can be generated, interactively analyzed and exported as svg.
Installation¶
For viewing pre-computed visualizations (available as .json files) no installation is required. This file can be imported on the demo website https://alitvteam.github.io/AliTV/d3/AliTV.html . If you are interested in creating whole genome alignment visualizations for your own data please refer to the README of the AliTV-perl-interface https://github.com/AliTVTeam/AliTV-perl-interface/ .
Citation¶
Please cite this article
when using AliTV. The source code in any specific version can additionally be cited using the zenodo doi, latest:
Tutorial I: Simple comparasion of chloroplast genomes¶
In this example chloroplast genomes of seven parasitic and non-parasitic plants are compared and analyzed with AliTV. The files needed for this tutorial are used as the default data, this means you need not import them.
Visualizing the Whole-Genome Alignment¶
Open
AliTV.htmlwith your favourite webbrowser. When you use AliTV the first time the chloroplast data are used as the default data.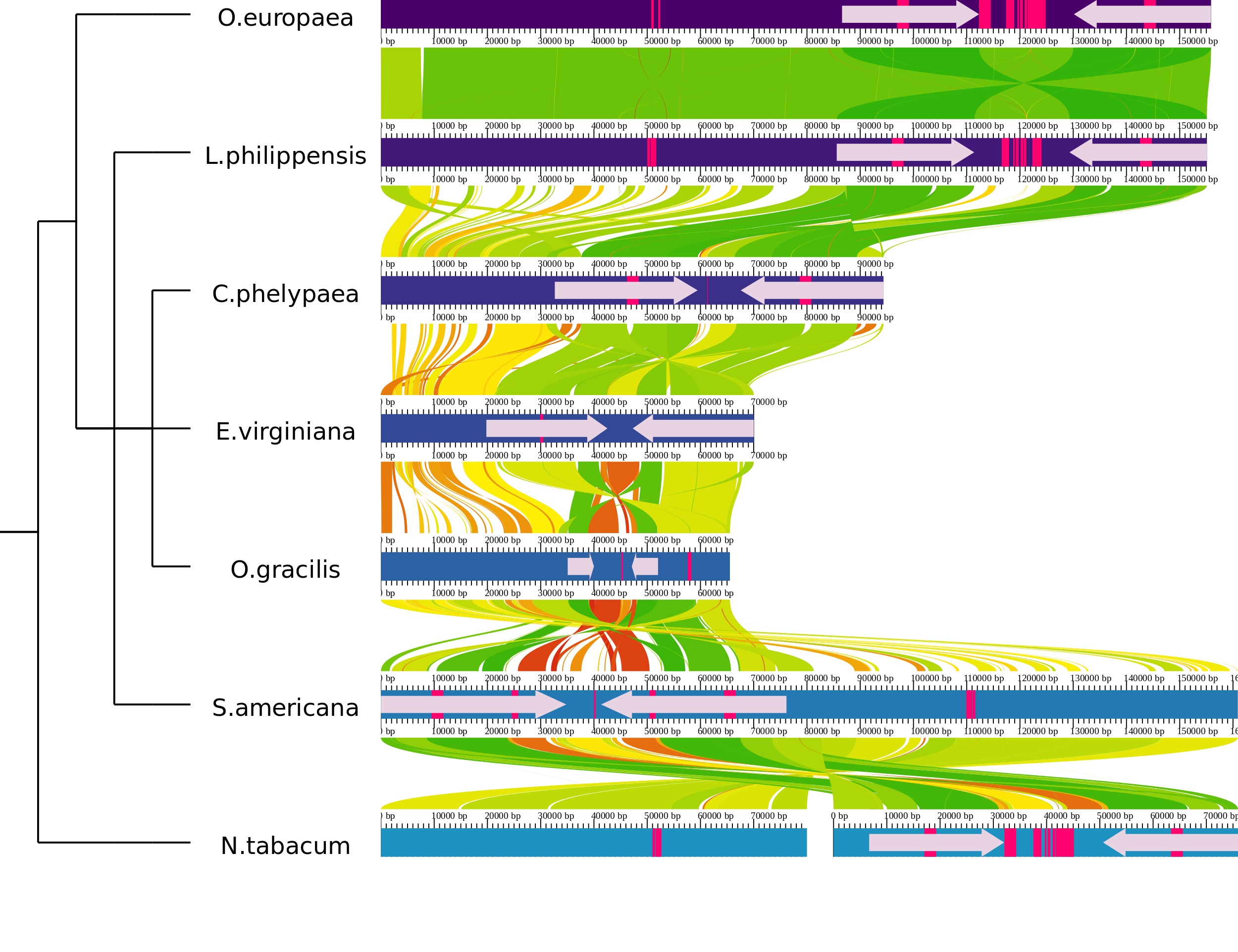
The AliTV image using the default configuration.
As you can see seven chloroplast genomes and the alignments between adjacent genomes are visualized. The genome of N. tabacum is splitted in two parts.
The phylogenetic tree represents the relationship between the analyzed genomes.
Create figure¶
- For using the figure you have to download it. Click for saving the picture.
- Open the folder where you saved the AliTV image and open it with your favourite image-viewer.
Change the genome orientation¶
- The direction of S. americana is the opposite in contrast to the adjacent genomes, because the alignments between them are twisted. Therefore AliTV provides the option to set the sequence orientation forward or reverse in order to obtain a clearer comparasion.
- A context menu appears by right-clicking the genome of S. americana. Select .
- With one easy mouseclick you have changed the orientation of S. americana and can now analyse the alignments between this and the adjacent genomes.
Set tree configurations¶
- In the current settings the phylogenetic tree of the analyzed plants is presented. AliTV provides easy options for changing the tree layout.
- Choose the tab on the HTML page.
- Here you can decide wether the tree is drawn or not. For the next few steps it is easier if the tree is not shown. Therefore make the checkbox unchecked.
- Submit the changes with
- Now the phylogenetic tree is hidden. You can easy show him by checking the checkbox again.
Change the genome and chromosome order¶
- If you want to compare N. tabacum to L. philippensis AliTV provides an easy option for changing the genome order.
- You can change the order by right-clicking N. tabacum. The context menu appears. Select . Now N. tabacum swapped its position with the genome below. So you can easy compare N. tabacum with L. philippensis
- For changing the order of chromosomes within a genome (for example N. tabacum) you select the chromosome which you want to resort. In the contextmenu select or .
- When you want to save the image with the current settings you can download it as described in Create Figure.
Filter links by identity and length¶
- For a biological analysis it may be helpful to filter links by their identity or length. AliTV offers both options for analyzing the image easy and interactive.
- Choose the tab on the HTML page.
- For filtering links you use the sliders. Set the range of the identity slider on 85% to 100%. Submit your changes with .
- As you can see some of the red and orange colored links are not shown because their identity is less than 85% and so they are filtered.
- In the same way links can be filtered by their length. So try it and have fun with this nice sliders!
Change graphical parameters¶
AliTV provides many ways to customise the image. The following list only shows a few examples. For more information checkout Features of AliTV or try it by yourself.
Setting the layout¶
- Select and choose your favourite layout (circular or linear).
- At the moment the circular layout is not the development stage of the linear layout. Therefore the most options and interactive functions are not working if you use it.
Coloring the chromosomes¶
With AliTV it is possible to change the color range of the presented genomes, the color of features and labels.
Select and define a new start and end color by using the color picker. It is also possible to type in the Hex or RGB value of your favourite color.
If you use #00ffc2 for color 1 and #ff8a00 for color 2 you get the following crazy AliTV image.

Setting new values for the genome color.
Scaling the chromosomes¶
If you want to change the default scaling of the sequences select and type in a new tick distance in bp.
The labeling of the ticks is changing as well, because every tenth tick is labeled by default. When you want to change the tick labels you can type in their frequency in the current tab.
If you set the tick distance to 10000bp and the label frequency to 3 you get the following image.

AliTV offers easy scaling of chromosomes.
Features of AliTV¶
Main Screen¶
Above is the user interface of AliTV available on the HTML page when you generate the figure.

User interface of AliTV
Contains information of the software as well as direct links to the demo version, the manual and the code documentation.
Will filter the alignments according to their identity and length by using the sliders. With the changes are submitted.
On top of that this tab contains the information about all links, features or chromosomes which are hidden in the current settings. By using the selectors you can show a specifc hidden element again. With clicking the changes are submitted.
AliTV provides the possibility to show genes, inverted repeats, repeats and N-stretches by default. If you assigned the necessary data to AliTV you can configurate them by using this feature.
With the checkboxes genes are shown, hidden or labeled. You can choose between a rect or an arrow and you can color them by using the colorpicker. With you submit the changes.
It is the same procedure for inverted repeats, repeats and N-stretches. But it is important that you assign the data to AliTV. Otherwise no biolgoical features are visualized.
With AliTV it is possible to visualize custom features like specific gene groups, t-RNAs and other crazy stuff you want to show on the chromosomes.
Therefore you can type in the name of your custom feature group, select a form and choose a color. That’s it! As you can see it is very easy to visualize every biological stuff with AliTV.
Here you can decide wether the phylogenetic tree is drawn or not. Moreover you can show the tree left or right to the alignments and you can change its width.
All parameters that deal with color, size and layout of the AliTV image can be setted here. You can change the image size, the colors of chromosomes and links and the labels.
AliTV uses the JSONEditor in order to offer you the possibility for
changing parameters, filters and data structure directly next to the
figure. By clicking the editor appears at the bottom of the HTML page.
First you see the structure of an AliTV object with data, filters
and conf. data contains the data and it simply consists of
karyo, links, features (optional) and tree (optional). All
graphical parameters like color, size, layout, etc. and other
configuration for drawing the AliTV image are written in conf.
Specific configurations like hidden chromosomes, links or features and
the minimal and maximal link identity and length are assigned to
filters. All modifications you made are submitted by clicking .
For more information about the object structure and the means of the variables check out the documentation of AliTV. If you have problems with the JSONEditor the following link may be helpful for you: https://github.com/josdejong/jsoneditor/.
If you want to use the AliTV image you have to download it. With you can download the AliTV image as SVG and open it with your favourite image-viewer. Moreover you can export the current settings in the JSON format by selecting . It may be helpful if you cant to save the settings and use it some other time. Then you can import JSON data by clicking .
Generating json files with alitv.pl¶
In case you want to visualize your own data you need to generate a json file.
This can be done using the alitv.pl perl script.
It has a simple mode where you just call it with a bunch of fasta files.
In this case pairwise alignments are calculated using lastz and a json file with default settings is created.
For more advanced usage there is the possibility to supply a yml file with custom parameters.
Please consult the README of the AliTV-perl-interface project for more information:
https://github.com/AliTVTeam/AliTV-perl-interface
Here is an outline of the steps required to reproduce the demo data sets:
Chloroplasts¶
This dataset consists of the chloroplasts of seven parasitic and non-parasitic plants.
All of those are published at NCBI with accession numbers:
NC_025642.1, NC_001568.1, NC_022859.1, NC_001879.2, NC_013707.2, NC_023464.1, and NC_023115.1
Along with the fasta files genbank files with annotations are available.
So it is easy to extract the locations of ndh and ycf genes as well as the inverted repeat regions.
This data set is also used as test set in AliTV-perl-interface so best refer to
https://github.com/AliTVTeam/AliTV-perl-interface/tree/master/data/chloroset
and specifically the input.yml file there.
Bacteria¶
This dataset consists of four strains of Xanthomonas arboricola of which two are pathogenic and two are not (Cesbron S, Briand M, Essakhi S, et al. Comparative Genomics of Pathogenic and Nonpathogenic Strains of Xanthomonas arboricola Unveil Molecular and Evolutionary Events Linked to Pathoadaptation. Frontiers in Plant Science. 2015;6:1126. doi:10.3389/fpls.2015.01126.). Data is available for download from:
The xanthomonas.yml file has the following content:
---
genomes:
-
name: Xanthomonas arboricola pv. juglandis JZEF
sequence_files:
- JZEF01.1.fsa_nt
-
name: Xanthomonas arboricola pv. juglandis JZEG
sequence_files:
- JZEG01.1.fsa_nt
-
name: Xanthomonas arboricola JZEH
sequence_files:
- JZEH01.1.fsa_nt
-
name: Xanthomonas arboricola JZEI
sequence_files:
- JZEI01.1.fsa_nt
alignment:
program: lastz
parameter:
- "--format=maf"
- "--ambiguous=iupac"
- "--strand=both"
- "--notransition"
- "--step=20"
So the parameters for lastz are set a little less sensitive (compared to default settings) to reduce runtime.
The resulting json file is still >25MB in size and contains lots of very short links with low identity.
In order to have better performance in the visualization those can be filtered on the json level with alitv-filter.pl
The commands to create the final json are:
alitv.pl xanthomonas.yml --project xanthomonas
alitv-filter.pl --in xanthomonas.json --out xanthomonas_arboricola.json --min-link-len 1000 --min-link-id 60
Chromosome 4¶
To demonstrate the capability of visualizing whole mammalian size chromosomes we imported a pre-calculated alignment of human and chimp chromosome 4 from Ensembl. This three files have been downloaded:
- ftp://ftp.ensembl.org/pub/release-87/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.chromosome.4.fa.gz
- ftp://ftp.ensembl.org/pub/release-87/fasta/pan_troglodytes/dna/Pan_troglodytes.CHIMP2.1.4.dna.chromosome.4.fa.gz
- ftp://ftp.ensembl.org/pub/release-87/maf/ensembl-compara/pairwise_alignments/homo_sapiens.GRCh38.vs.pan_troglodytes.CHIMP2.1.4.lastz_net.tar
After unpacking all alignments those to chromosome 4 of human were combined.
Usually alitv.pl handles tasks like renaming ids to make them unique transparently.
However, this can not be done when importing pre-calculated alignments.
In our example both chromosomes have the id 4 and are prefixed in the maf file with homo_sapiens. and pan_troglodytes. respectively.
Furthermore the file chr4.maf contains all alignments to chromosome 4 of human and we only want those from chromosome 4 of chimp.
So to combine and clean the maf and fasta files and to prepare them for import into AliTV you can do the following:
zcat *H.sap.4.* >chr4.maf
perl -pe 's/homo_sapiens\.4/h4/;s/pan_troglodytes.4/p4/' chr4.maf >chr4.fixID.maf
perl -ne 'print if(/^#/);if(/^a/){$s=$_}if(/^s h4/){$h=$_}if(/^s p4/){print $s.$h.$_."\n"}' chr4.fixID.maf >chr4.clean.maf
perl -pe 's/^>4/>h4/' Homo_sapiens.GRCh38.dna.chromosome.4.fa >Homo_sapiens_chr4.fa
perl -pe 's/^>4/>h4/' Pan_troglodytes.CHIMP2.1.4.dna.chromosome.4.fa >Pan_troglodytes_chr4.fa
Now alitv.pl can be executed with the chr4.yml:
---
genomes:
-
name: Homo sapiens
sequence_files:
- Homo_sapiens_chr4.fa
-
name: Pan troglodytes
sequence_files:
- Pan_troglodytes_chr4.fa
alignment:
program: importer
parameter:
- "chr4.clean.maf"